Validation library
Coming soon
We're publishing the validation library one study at a time. This report is being finalized for release — check back soon.
← Back to the validation libraryWe're publishing the validation library one study at a time. This report is being finalized for release — check back soon.
← Back to the validation libraryAbstract. This is a two-sided positive control for Haritica's
Linkage Mapper, an independent reimplementation of the published MMAPPR method (Mutation Mapping Analysis Pipeline for Pooled RNA-seq), comparing Haritica's in-app cloud
output against an independent MMAPPR2 reference. The maize glossy13 (gl13)
dataset (Li et al. 2013; BioProject PRJNA622294) contains three independent EMS allele families,
each sequenced as a glossy-mutant pool versus a wild-type-sibling pool by RNA-seq — a classic
bulked-segregant RNA-seq (BSR-Seq) design. Reads were aligned to maize B73 NAM-5.0 and analysed with the
original MMAPPR2 R/Bioconductor package, run as a separate tool on an AWS instance. MMAPPR2 computes
a per-base Euclidean distance between the pools, raises it to the fourth power, fits a per-chromosome
AICc-optimized Loess regression, and localizes the linkage peak with a bootstrap confidence interval. In
all three families the reference peak falls on chromosome 3, the published location of the
causal gene gl13 = GRMZM2G118243 (an ABCG transporter validated by multiple
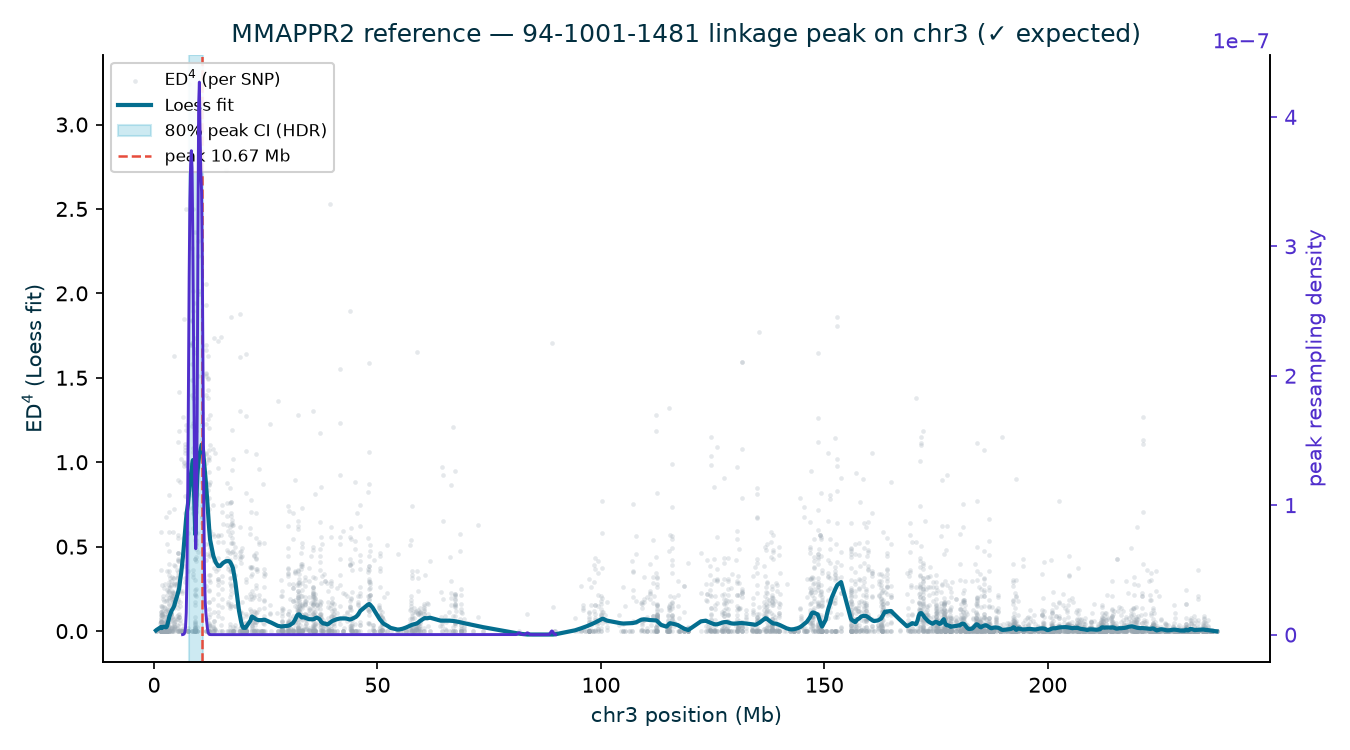
premature-stop EMS alleles and a knockout): N211B 5.84 Mb (CI 3.75–14.64), 94-1001-1481
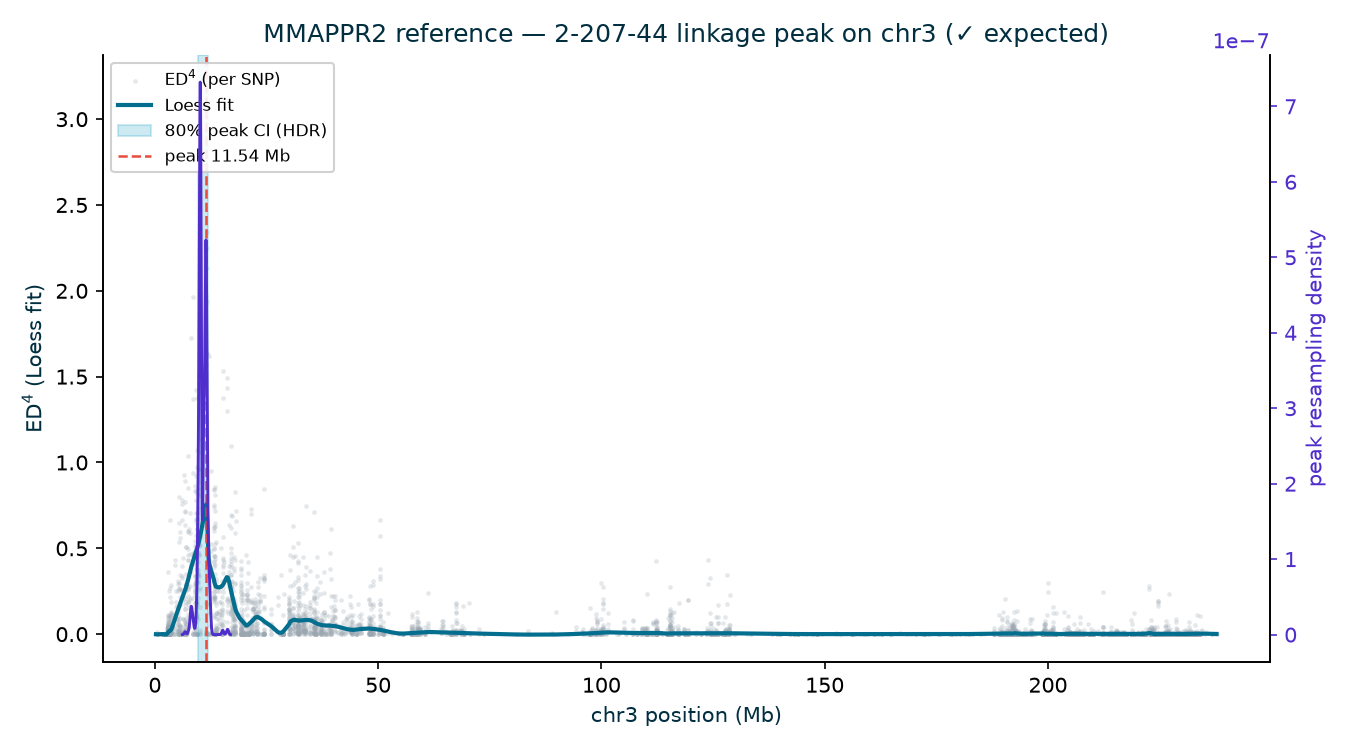
10.67 Mb (CI 7.73–10.95), 2-207-44 11.54 Mb (CI 9.69–11.81). Three independent recoveries of
the same chromosome make this a stringent positive control.
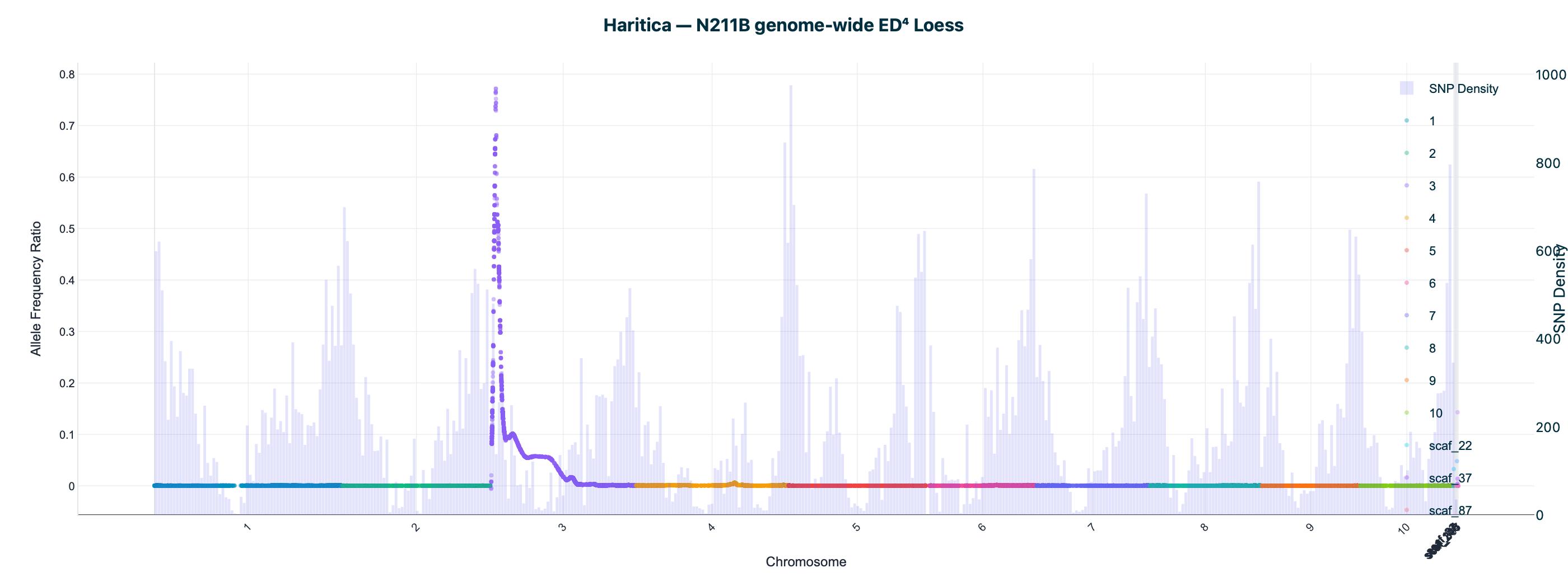
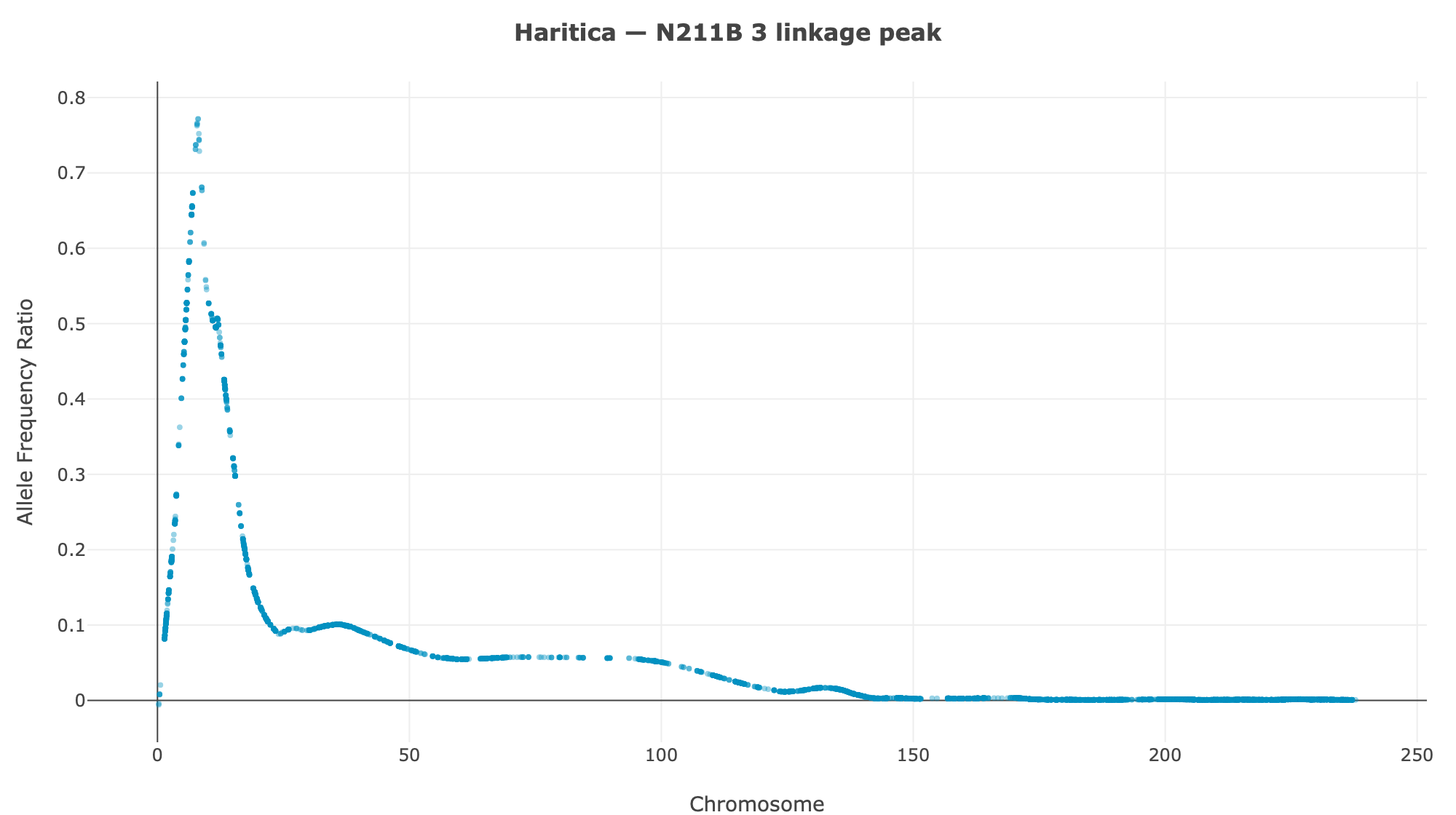
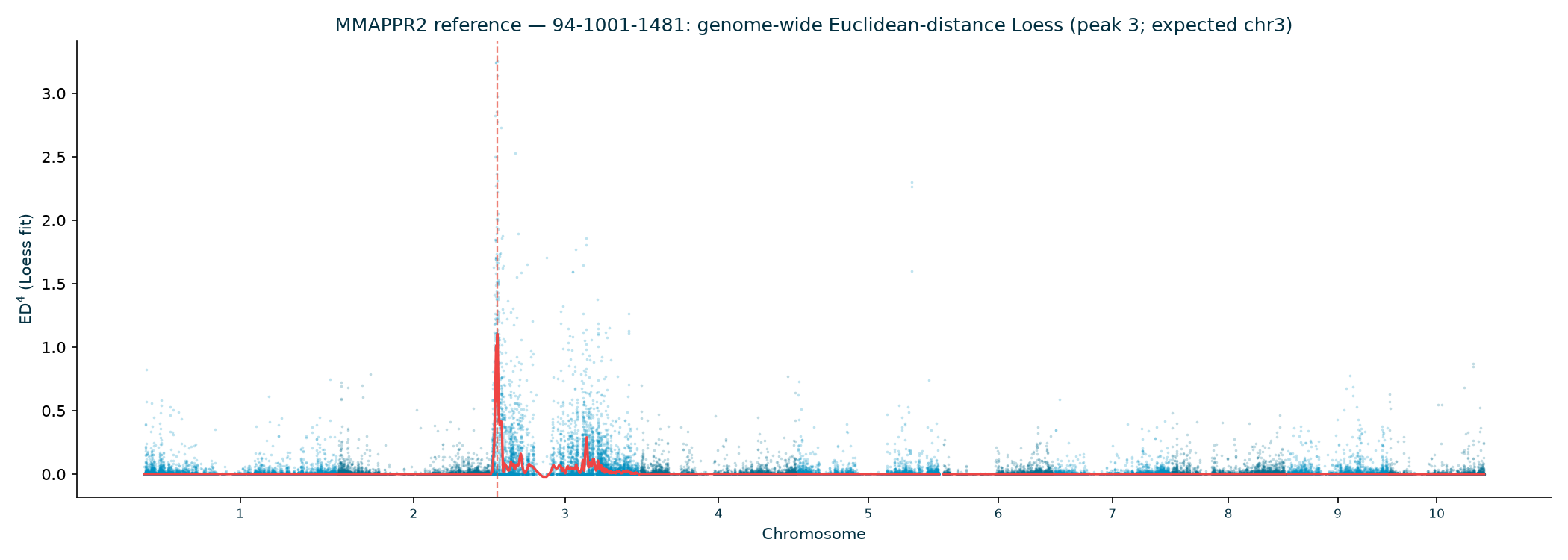
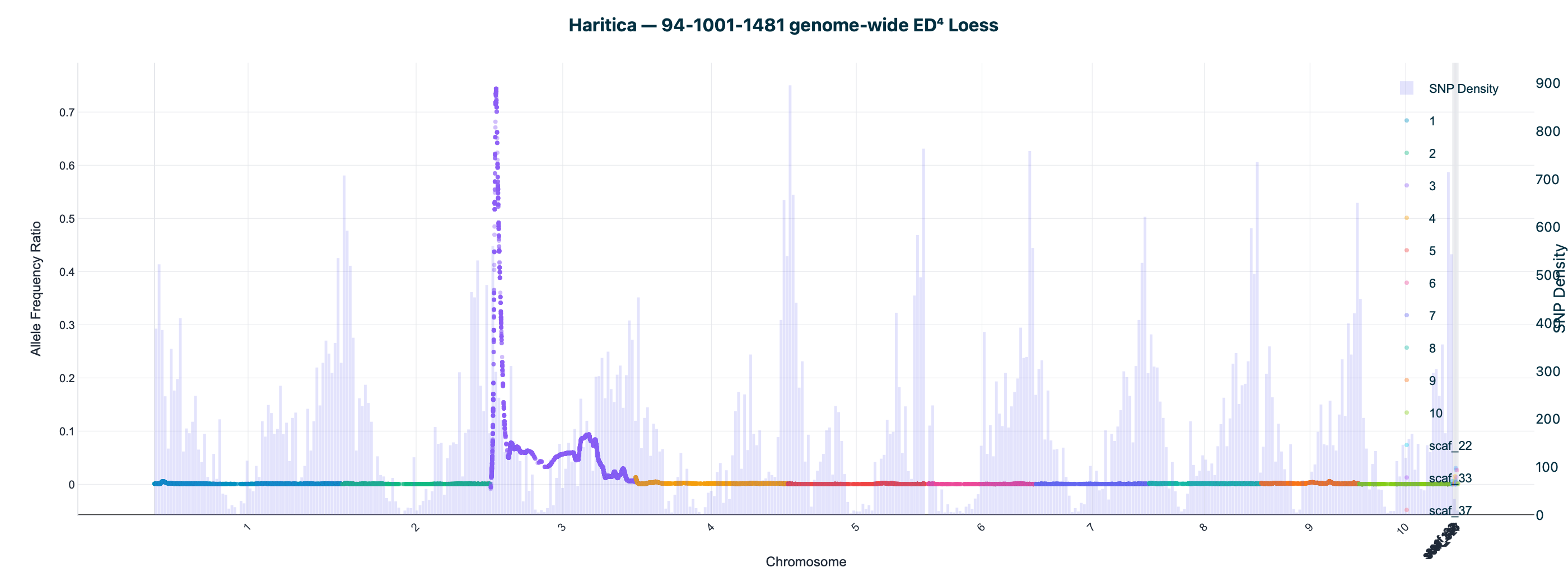
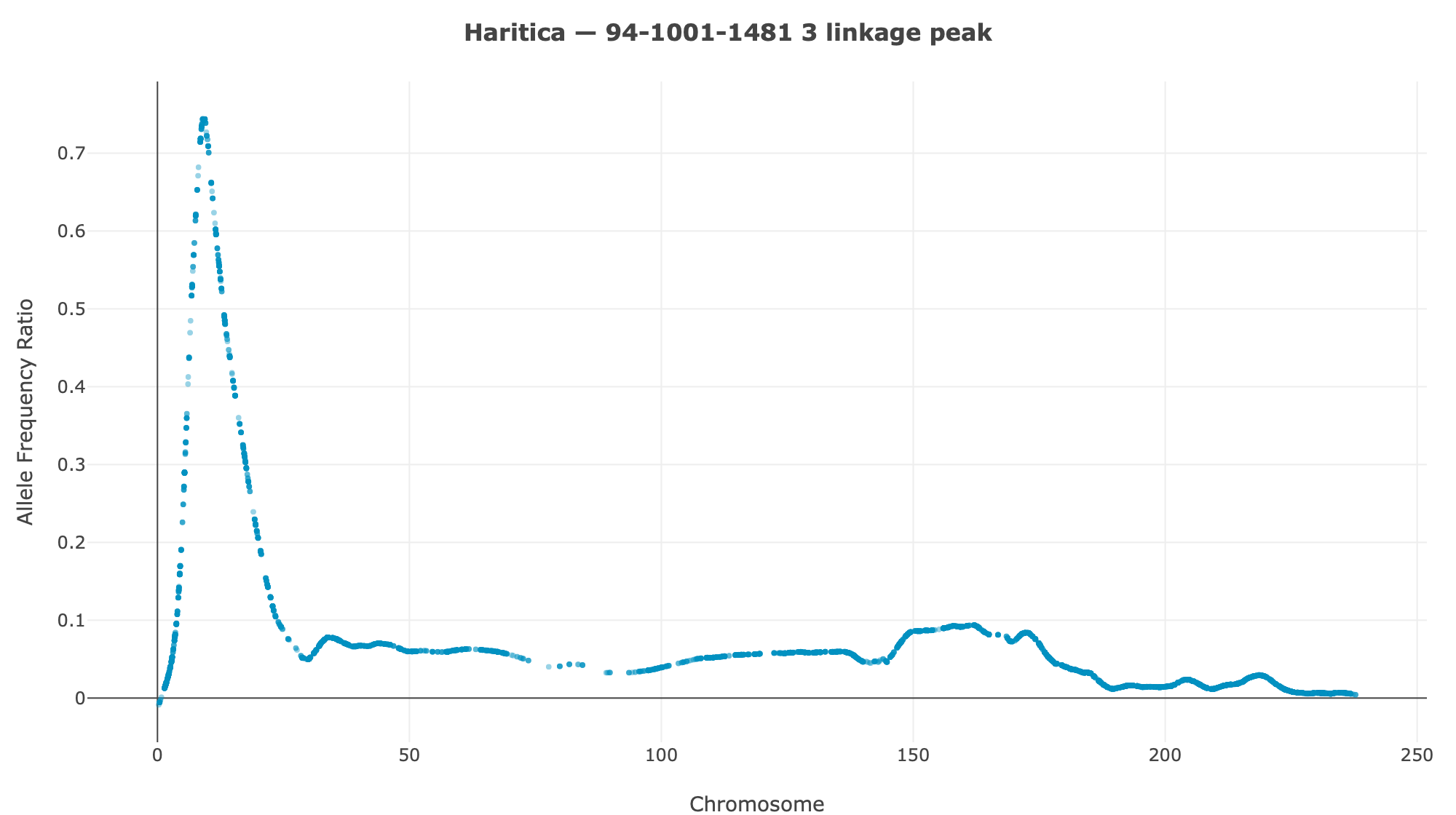
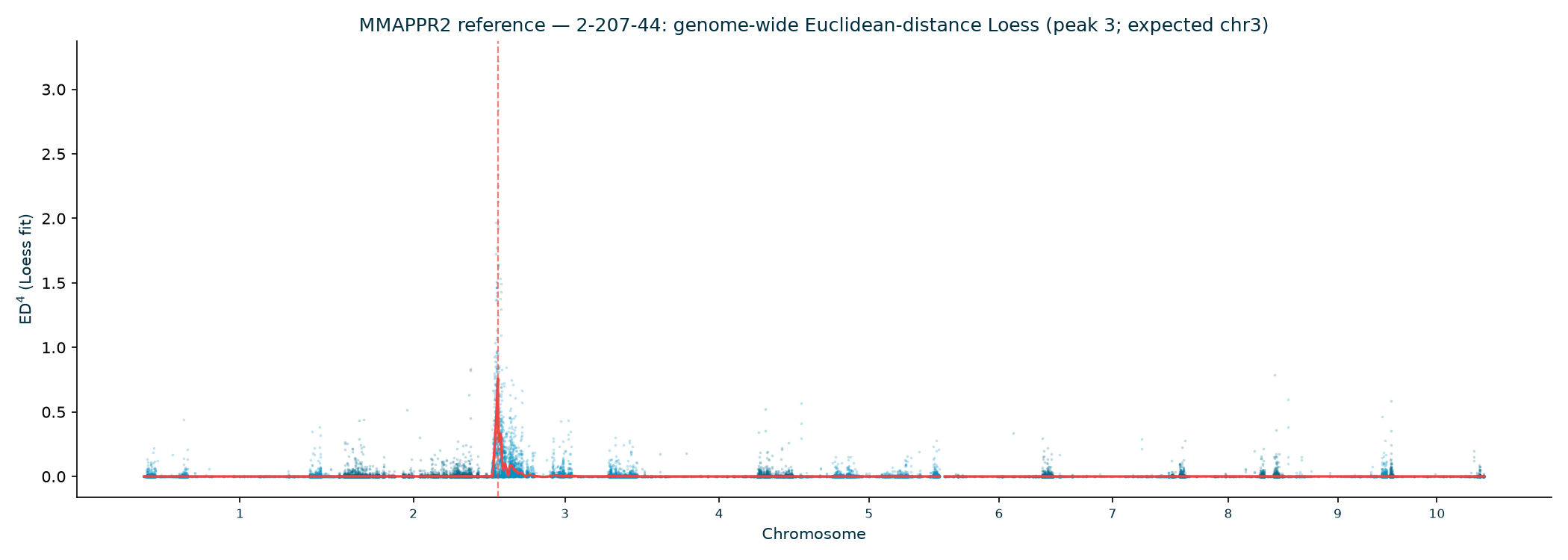
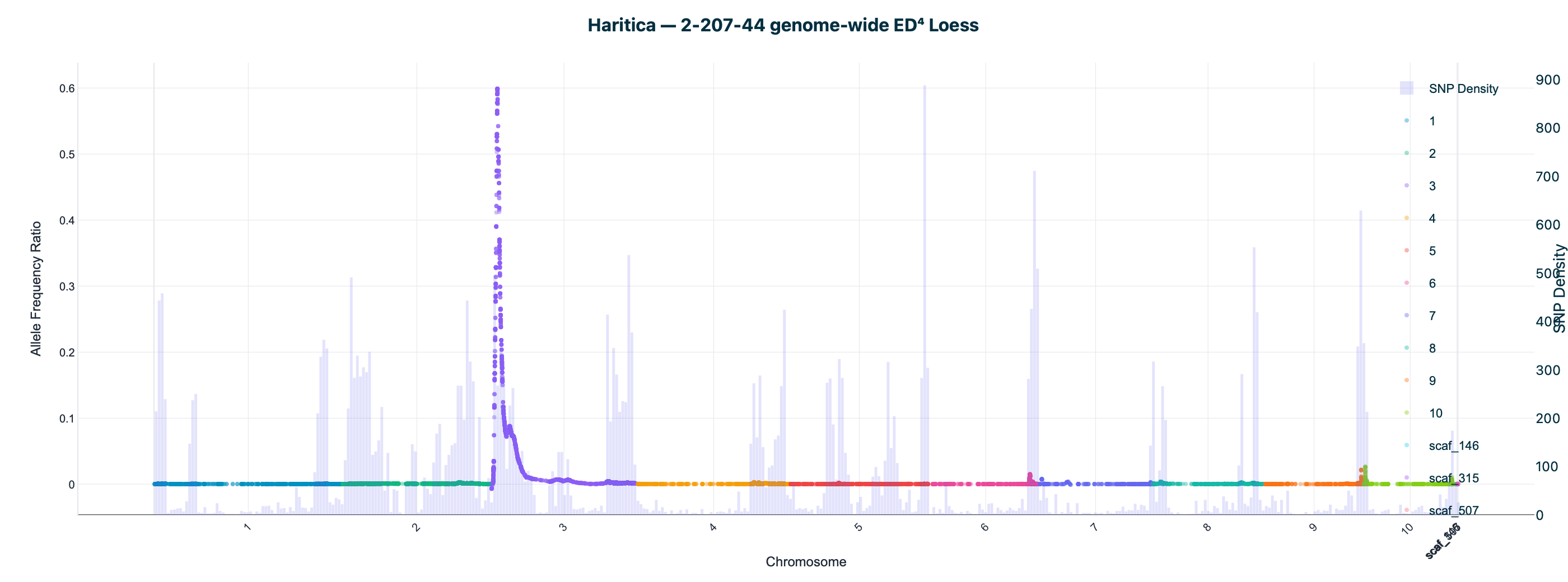
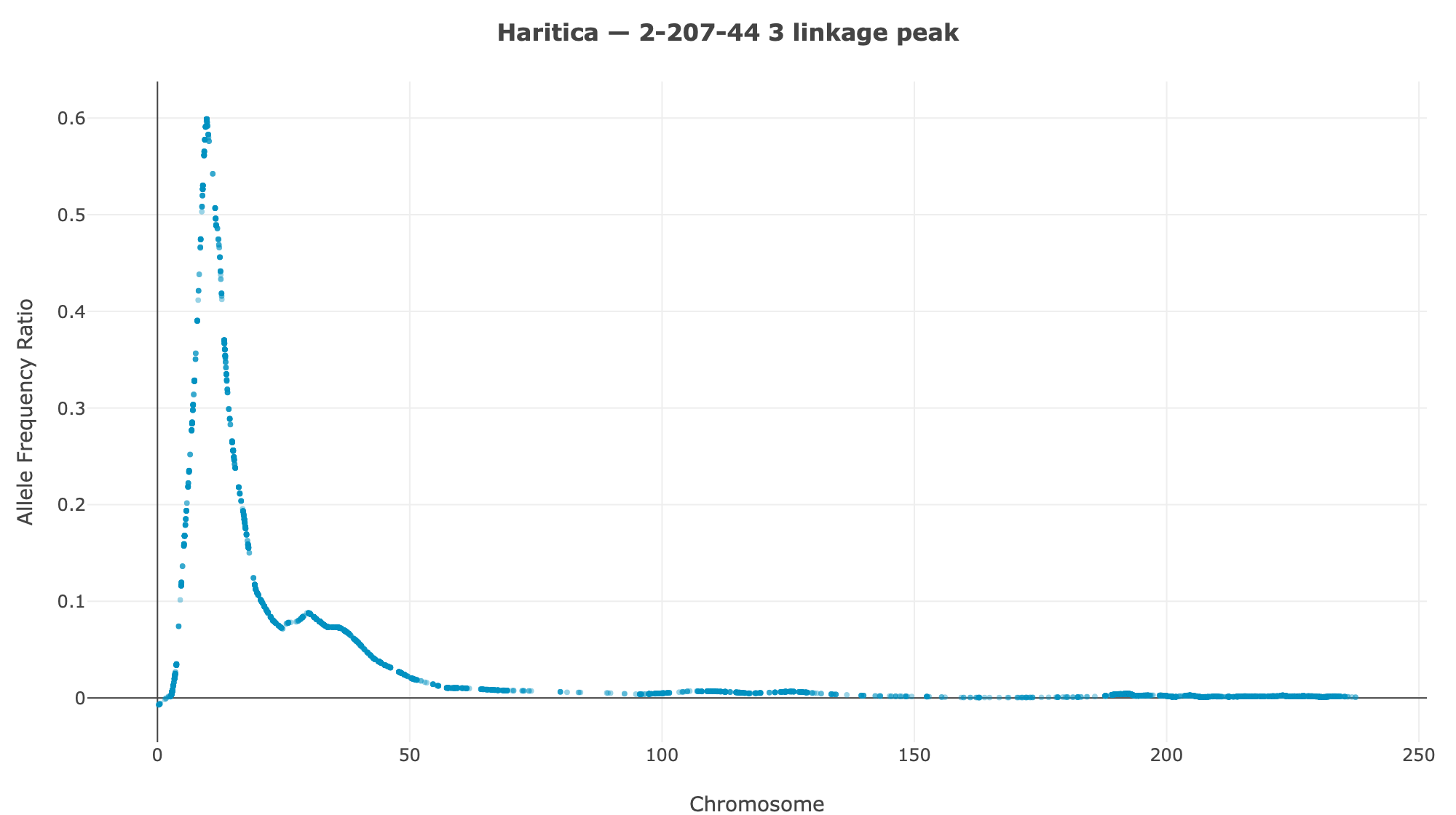
Haritica result. Haritica's Linkage Mapper, run in the cloud on the same three BAM sets with matched parameters, independently recovers chromosome 3 in all three families: N211B 8.06 Mb (ED⁴ 0.77, 87,283 SNPs), 94-1001-1481 9.00 Mb (0.74, 76,050), 2-207-44 9.75 Mb (0.60, 33,180). Every Haritica peak falls inside the reference's bootstrap CI, and the published gl13 locus (chr3:10.26–10.27 Mb) lies within the supported intervals — see the side-by-side figures and §2a.
The dataset (Li et al. 2013 [1]; BioProject
PRJNA622294) is the maize glossy13
BSR-Seq panel. gl13 was cloned by combining bulked-segregant RNA-seq with Seq-walking, and confirmed
as GRMZM2G118243 by multiple EMS alleles carrying premature stop codons plus a knockout allele.
Three independent EMS allele families (N211B, 94-1001-1481, 2-207-44) were each phenotypically sorted into a
glossy-mutant pool and a wild-type-sibling pool and RNA-sequenced (paired-end ~101 bp, Illumina;
Table 1). Reads were aligned to maize B73 NAM-5.0 (Ensembl Plants release-62) with HISAT2
(splice-aware paired-end; basic index built on the instance), sorted and indexed with samtools. MMAPPR2 then performed the linkage analysis,
independently, for each family.
| Allele family | Mutant pool | WT-sibling pool | Causal gene | Expected peak |
|---|---|---|---|---|
| N211B | SRR11457982 | SRR11457981 | gl13 = GRMZM2G118243(ABCG transporter) | chr 3 (AGPv2 ~10.27 Mb) |
| 94-1001-1481 | SRR11457984 | SRR11457983 | ||
| 2-207-44 | SRR11457986 | SRR11457985 |
| Parameter | Value |
|---|---|
| Reference tool | MMAPPR2 (jonathonthill, commit 37a5d00), GPL-3 |
| Aligner | HISAT2 2.2.1 → B73 NAM-5.0 (basic index built on instance), sorted/indexed BAM |
| Variant tally | Rsamtools::pileup (simpleCigar=TRUE; junction reads excluded) |
| Distance metric | per-base Euclidean distance over [A,C,G,T] pool frequencies, raised to distancePower |
| distancePower | 4 |
| minDepth / homozygoteCutoff | 20 / 0.95 |
| minBaseQuality / minMapQuality | 20 / 20 |
| Loess | per-chromosome, AICc-optimized span (loessOptResolution 0.001) |
| Peak refinement | 1000-replicate 50% subsample → KDE of peak position → highest-density region |
| peakIntervalWidth | 0.80 |
The reference is MMAPPR2 [2,3], the R/Bioconductor implementation of the MMAPPR algorithm
(Hill et al. 2013 [2]), run by a thin driver (run_reference_mmappr2.R)
that calls the package and adds no statistics of its own. This control is independent on three axes:
(1) the data is third-party — the gl13 study (Schnable lab) produced PRJNA622294, not
Haritica and not the MMAPPR2 authors; (2) the reference is a separate codebase that does its own
variant calling (Rsamtools pileup) on the BAMs, then its own Loess and bootstrap; (3) the biology is an
external published result — gl13 was positionally cloned to GRMZM2G118243 on
chromosome 3 and confirmed by independent alleles, so “the peak lands on chr 3 over
gl13” is a blind test. The design also gives a fourth layer: the same locus is recovered in
three independent EMS families, so a chance hit is implausible (≈(1/10)3 for ten maize
chromosomes).
Limits. The reference isolates the variant-calling + distance + Loess + peak stages from the alignment step (held common because the Haritica run uses the same BAMs); it is not an end-to-end custody audit. The published gl13 interval is in AGPv2 coordinates while this run aligns to B73 NAM-5.0 — chromosome assignment (chr 3) is build-robust, and the exact v5 coordinate of the gl13 model is read from MMAPPR2’s candidate table for the chr 3 peak. It is one study, run by us; independence is methodological (third-party data + a separate standard package + published biology), not custodial.
MMAPPR2 was run on a 64-vCPU AWS EC2 instance (m6i.16xlarge, on-demand; MMAPPR2 is computationally expensive
— genome-wide pileup on the 2.4 Gb maize genome, per-chromosome AICc-Loess, and a 1000-replicate
bootstrap — for three families in parallel). Each reference figure is rendered from MMAPPR2’s
own computed output (its per-position distance/Loess values and the peak/CI/resampling density);
no science is recomputed downstream. Native MMAPPR2 plots and clean re-renders (styled to match
Haritica’s Loess view for the side-by-side comparison) were produced by
make_reference_figures.py.
MMAPPR2 is GPL-3; minimap2 is MIT. MMAPPR2 is run here only as a separate tool to generate the reference; its outputs are facts and are not bundled or ported into closed-source Haritica — the same posture as the differential-expression control running GPL R DESeq2. Haritica's Linkage Mapper, shown alongside, is a clean-room reimplementation of the published method, not a translation of GPL-3 source.
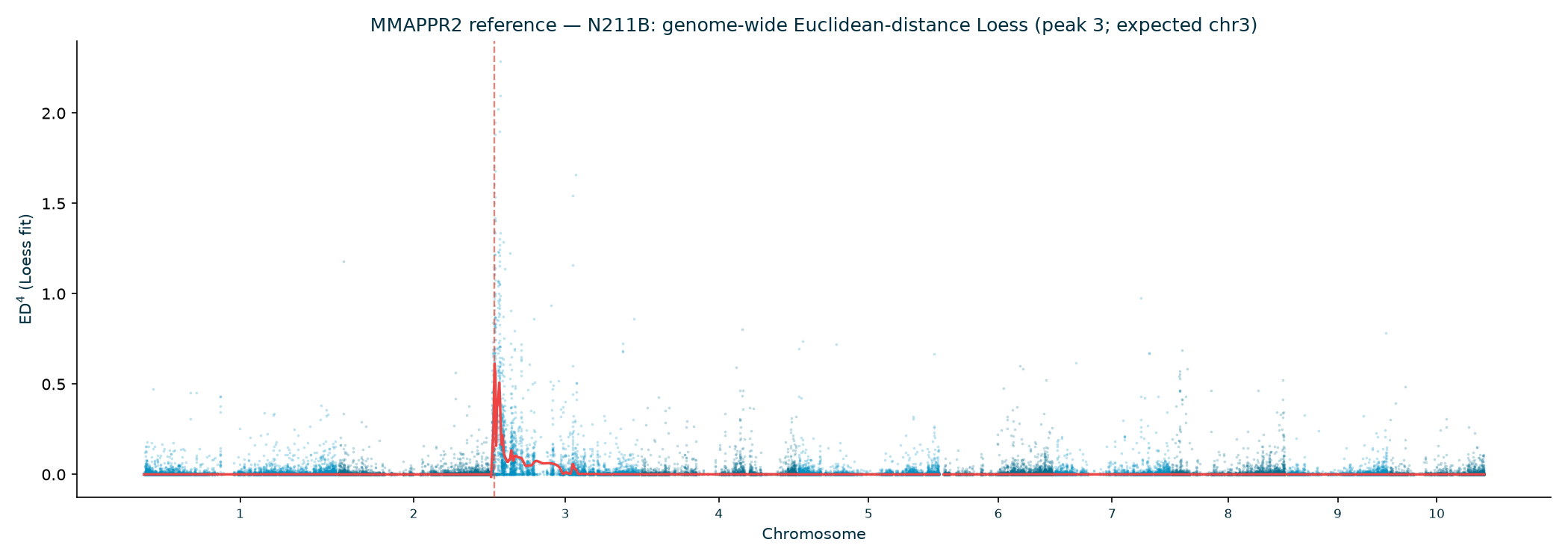
In every allele family the genome-wide Euclidean-distance Loess has a single dominant peak on
chromosome 3, and the bootstrap confidence interval brackets the gl13 gene (Table 3;
Figures 1–6). The peak is corroborated by a source we do not produce (Table 4): the
independent cloning of gl13 to GRMZM2G118243 on chr 3.
Most strikingly, the reference recovers the gene itself — blind. MMAPPR2’s candidate
table for the chr 3 peak flags, in two independent families, distinct EMS premature-stop codons
in the same gene model Zm00001eb122470 (= gl13, chr3:10,262,628–10,272,956):
a Q→* at 10,266,222 (N211B) and a W→* at 10,267,605 (2-207-44). This reproduces the paper’s
allelic premature-stop validation without ever being told the answer, and pins a v5 coordinate that matches
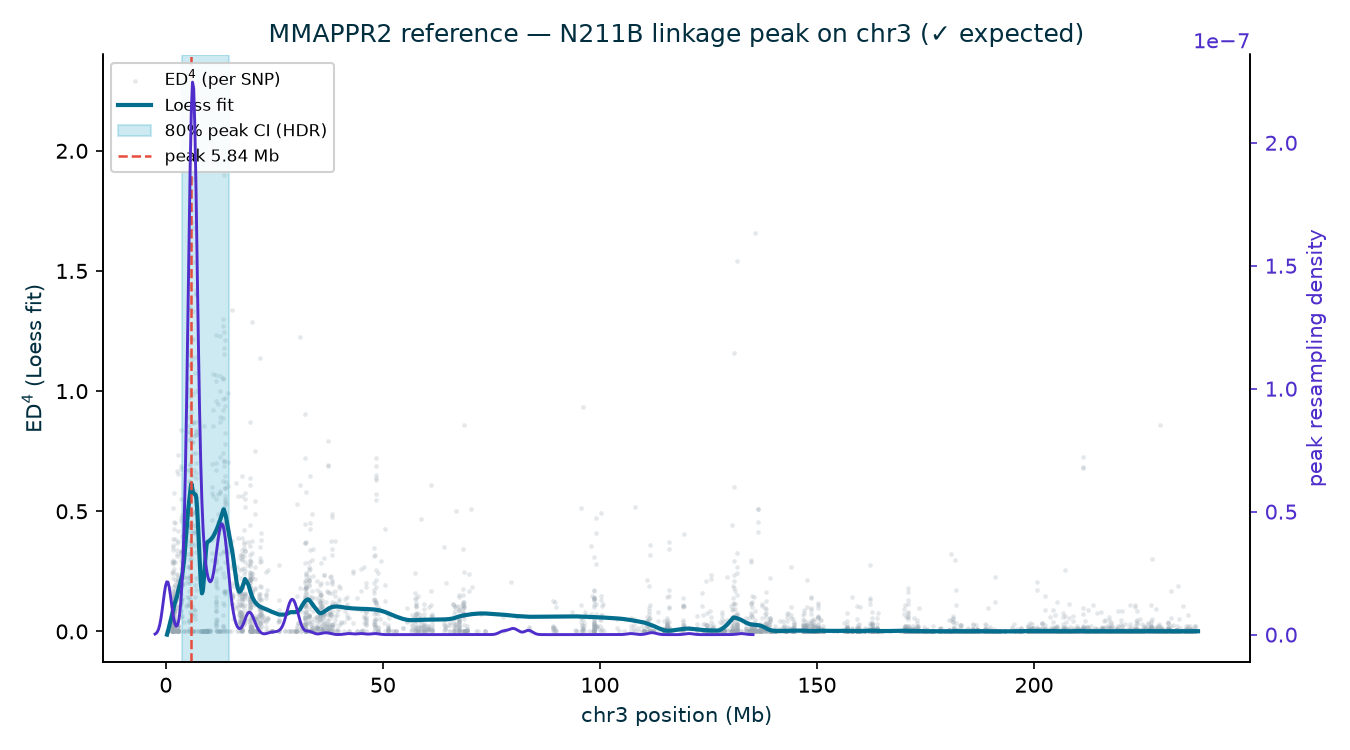
the published AGPv2 position to within ~6 kb. (The N211B Loess argmax sits at 5.84 Mb, but its wide
80% interval, 3.75–14.64 Mb, brackets gl13; the deeper 94-1001-1481 and 2-207-44 families
localize to 10.7 and 11.5 Mb with tighter intervals.)
| Family | Peak chr | Peak (Mb) | ED⁴ (Loess) | 80% CI (Mb) | Hit chr3? |
|---|---|---|---|---|---|
| N211B | 3 | 5.84 | 0.614 | 3.75–14.64 | ✓ |
| 94-1001-1481 | 3 | 10.67 | 1.104 | 7.73–10.95 | ✓ |
| 2-207-44 | 3 | 11.54 | 0.759 | 9.69–11.81 | ✓ |
| Source | Result |
|---|---|
| Published cloning (Li et al. 2013) | gl13 = GRMZM2G118243, chromosome 3 (AGPv2 ~10.27 Mb); confirmed by multiple EMS premature-stop alleles + a knockout |
| MMAPPR2 candidate table (this run, v5) | gl13 gene model in the chr 3 peak interval: Zm00001eb122470 = gl13, chr3:10.26–10.27 Mb (independent EMS premature-stop alleles recovered: N211B Q→* @10.266 Mb, 2-207-44 W→* @10.268 Mb) |
| MMAPPR2 peak chromosome (all 3 families) | 3 / 3 / 3 |
MMAPPR2 — reference
Haritica
MMAPPR2 — reference
Haritica
MMAPPR2 — reference
Haritica
MMAPPR2 — reference
Haritica
MMAPPR2 — reference
Haritica
MMAPPR2 — reference
Haritica
MMAPPR2’s native genome/peak plots are included as
provenance under figures/ref_<family>_native_*.png.
Both pipelines were run on the same three HISAT2-aligned BAM sets with matched parameters (ED power 4, homozygote cutoff 0.95, min depth 20, min base/map quality 20). Haritica's Linkage Mapper was run in the cloud (AWS Batch); its genome-wide and chr 3 plots above are real in-app Plotly renderings of the job output. The reference is the independent MMAPPR2 package.
| Family | Haritica peak | Haritica ED⁴ | SNPs | MMAPPR2 peak | MMAPPR2 80% CI | Same chr | Haritica peak ∈ ref CI |
|---|---|---|---|---|---|---|---|
| N211B | chr3 · 8.06 Mb | 0.77 | 87,283 | chr3 · 5.84 Mb | 3.75–14.64 Mb | ✓ | ✓ |
| 94-1001-1481 | chr3 · 9.00 Mb | 0.74 | 76,050 | chr3 · 10.67 Mb | 7.73–10.95 Mb | ✓ | ✓ |
| 2-207-44 | chr3 · 9.75 Mb | 0.60 | 33,180 | chr3 · 11.54 Mb | 9.69–11.81 Mb | ✓ | ✓ |
Verdict: PASS. Haritica's Linkage Mapper independently recovers chromosome 3 in all three
EMS families, the published location of gl13 (GRMZM2G118243 = Zm00001eb122470,
chr3:10.26–10.27 Mb). Every Haritica peak falls inside the reference's 80% bootstrap CI, and the
causal locus lies within the supported intervals. Exact peak positions and ED⁴ magnitudes differ modestly
between the two — expected, since even on identical BAMs Haritica's per-base allele counting, SNP filtering
and AICc-Loess are an independent reimplementation of the MMAPPR method, not a port. The
positive-control criterion (the chromosome-level linkage call, with CI overlap and recovery of the published
gene) is met by all three families.
All inputs are public. Raw reads: BioProject PRJNA622294 (ENA), runs SRR11457981–SRR11457986. Reference genome: maize B73 Zm-B73-REFERENCE-NAM-5.0, Ensembl Plants release-62 (FASTA + GTF). The reference is generated by MMAPPR2 in a Bioconductor Docker image and rendered to figures; the Haritica side is its in-app cloud Linkage Mapper. Exact parameters: reference_parameters.json.
37a5d00, GPL-3.Reference side. Reproduced end-to-end on a 64-vCPU AWS instance: download the public reads → HISAT2 align to B73 NAM-5.0 → MMAPPR2 ×3 families → render reference figures from MMAPPR2's own computed output. The shared maize BAMs are archived in S3 so the Haritica run uses the identical alignments.
Haritica side. The three BAM sets were staged to the user's cloud input prefix and submitted to
the Haritica Linkage Mapper in BAM mode, in the cloud (AWS Batch), with the matched parameters (ED
power 4, homozygote cutoff 0.95, min depth 20, min base/map quality 20 — full list
in reference_parameters.json). Each cloud job's per-SNP output
(mmappr_results.csv) was rendered by the live Haritica app and captured as a clean Plotly PNG
(Plotly.toImage, not a screenshot). All three families returned the chromosome 3 peak
blind — see §2a.
Note on cloud performance. The maize genome-wide
pileup is large; Haritica's Linkage Mapper backend parallelizes it across genomic windows (independent
samtools mpileup -r per window, mirroring MMAPPR2's per-chromosome Rsamtools approach), which
brings a whole-genome RNA-seq pileup down to a few minutes on the cloud worker.