Positive control · SRR14253270 · Homo sapiens, GM12878 / HG001 RNA-seq
Validation of Haritica variant annotation against the NIST Genome-in-a-Bottle HG001 truth set
Haritica (cyvcf2 / SnpEff) versus the NIST GIAB HG001 GRCh38 v4.2.1 benchmark and the independent bcftools statistics
Dataset SRR14253270 (GM12878 / HG001) · references NIST GIAB v4.2.1 (Zook et al. 2019), bcftools 1.x (Danecek et al. 2021), SnpEff (Cingolani et al. 2012) · Homo sapiens GRCh38, Ensembl 110, chr20–21 · 2026
Abstract. A published human RNA-seq run (SRR14253270, a GM12878 sample — the same individual,
HG001/NA12878, behind the NIST Genome-in-a-Bottle reference) was processed end-to-end in Haritica
— HISAT2 alignment, bcftools calling, and SnpEff annotation on chromosomes 20 and 21
— yielding 610 quality-passed variants (540 SNVs, 70 indels). Two independent anchors validate the
result. Against the externally-published NIST GIAB HG001 GRCh38 v4.2.1 small-variant truth set, restricted
to its high-confidence regions, the calls reach precision 0.53 (265 true positives, 235 false positives),
rising to 0.62 once the ADAR A>G / T>C RNA-editing signature — real RNA biology absent from a DNA
truth set — is set aside. Independently, Haritica computes the Ti/Tv ratio, the 12-class substitution
spectrum, and the depth and quality histograms in its own Python (cyvcf2), and bcftools computes
the same quantities in C: the two agree bit-for-bit (Ti/Tv 3.06 = 3.06; all twelve substitution
classes identical). SnpEff's consequence calls agree with bcftools csq on 66.8% of variants,
rising to 97.1% on the consequence classes both tools can emit.
1Data and methods
The input is a single published Illumina paired-end RNA-seq run, SRR14253270, from GM12878 — the
lymphoblastoid cell line of individual NA12878, which NIST designates HG001 and characterises in its
Genome-in-a-Bottle reference materials. Within Haritica, reads were aligned to Ensembl GRCh38 (release 110)
with HISAT2, restricted to chromosomes 20 and 21, called with bcftools mpileup
(-q20 -Q20 -B) followed by bcftools call -mv, and annotated with SnpEff
(GRCh38.99). Of 2,882 raw variants, 610 passed quality control (540 SNVs, 70 indels; Ti/Tv 3.06).
HISAT2 is used because the data are short-read RNA-seq; the minimap2 default -ax splice preset
targets long reads.
Table 1. Analysis parameters for the Haritica run.
Parameter
Value
Input
GM12878 RNA-seq SRR14253270, Illumina paired-end, 8 M read pairs
Reference genome
Ensembl GRCh38 (release 110), prebuilt HISAT2 index
Chromosomes
chr20, chr21
Aligner
HISAT2 (short-read spliced)
Variant calling
bcftools mpileup -q20 -Q20 -B → call -mv
Annotation
SnpEff GRCh38.99
Truth set
GIAB HG001_GRCh38_1_22_v4.2.1_benchmark VCF + high-confidence BED
Result
610 passed (540 SNV / 70 indel); Ti/Tv 3.06
1.1 Accuracy against an external truth set
The variant calls are scored against the NIST Genome-in-a-Bottle HG001 / NA12878 GRCh38 v4.2.1
small-variant benchmark [1], the canonical truth set for human variant calling, which Haritica did not
produce. Concordance is measured with standard tooling — bcftools norm followed by
bcftools isec, restricted to the benchmark's high-confidence regions — by the script
reference_concordance.py. Because the input is a real,
published RNA-seq run rather than simulated data, the comparison reflects a true end-to-end pipeline run.
The truth set is whole-genome DNA, so region-level recall is low by construction (a single RNA-seq run
samples only expressed exons); the informative quantity here is precision — what fraction of the
pipeline's calls are real.
1.2 Independent cross-check of the statistics
Haritica computes the Ti/Tv ratio, the 12-class substitution spectrum, and the depth and quality
histograms in its own Python, parsing the VCF with cyvcf2. bcftools [2] computes the same
quantities independently in C. Bit-identical agreement therefore means two separate codebases, in two
languages, reach exactly the same answer on the same file; an exact integer match across all twelve
substitution classes is not attributable to a shared coincidental error. SnpEff's [3] consequence calls
are corroborated in the same spirit: bcftools csq annotates the identical VCF and the two
assignments are compared per variant by annotation_concordance.py.
One caveat, stated plainly: cyvcf2 uses HTSlib for low-level VCF parsing, as bcftools does,
so the independence is at the statistics and algorithm layer — which is the layer these figures
validate.
2Plot validation
The substitution, depth, indel, and quality views read the pipeline's output VCF and are compared
against the same quantities computed independently by bcftools stats / plot-vcfstats
(Figures 1–4). These pairs validate the statistics and rendering rather than the calling itself;
the proof that the pipeline recovers real biology is the GIAB concordance in §3. The substitution
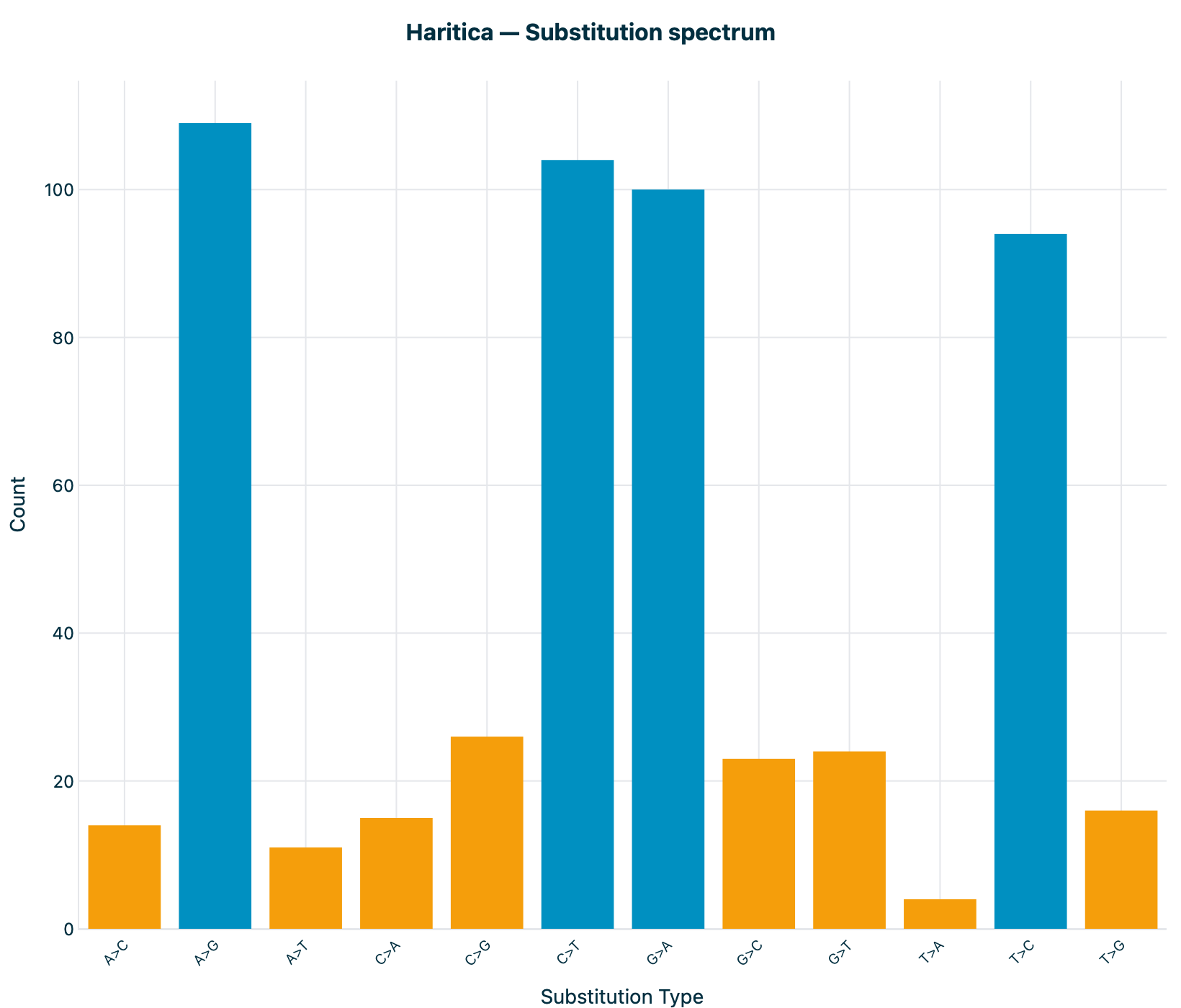
spectrum is bit-identical (A>G 109, C>T 104, G>A 100, T>C 94, …), with
the four transition bars dominating (Ti/Tv 3.06, elevated by coding SNVs and the ADAR A>G / T>C RNA-editing
signature). The impact and consequence distributions (Figures 5–6) render SnpEff's tiers and effect
table; SnpEff's native charts depend on the legacy Google Charts API, so there is no permissive plot to
overlay and the annotation engine is instead validated against bcftools csq (§4). The
oncoplot and rainfall views (Figure 7) are maftools-style visualizations shown for completeness:
maftools (GPL R) is the only canonical source and is excluded for licensing, and these are single-sample.
Haritica
bcftools plot-vcfstats — reference
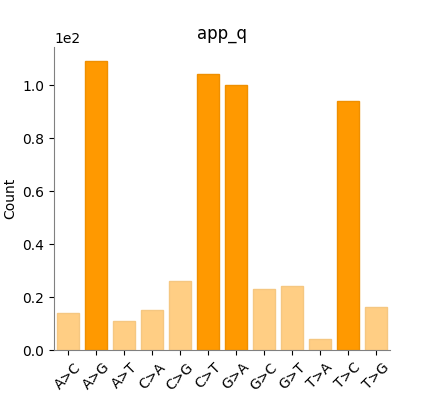
Figure 1. Substitution spectrum (12 classes). Counts are bit-identical
between Haritica's cyvcf2 statistics and bcftools' plot-vcfstats (MIT): A>G 109,
C>T 104, G>A 100, T>C 94, …. The four transition bars dominate
(Ti/Tv 3.06).
Haritica
bcftools stats — reference
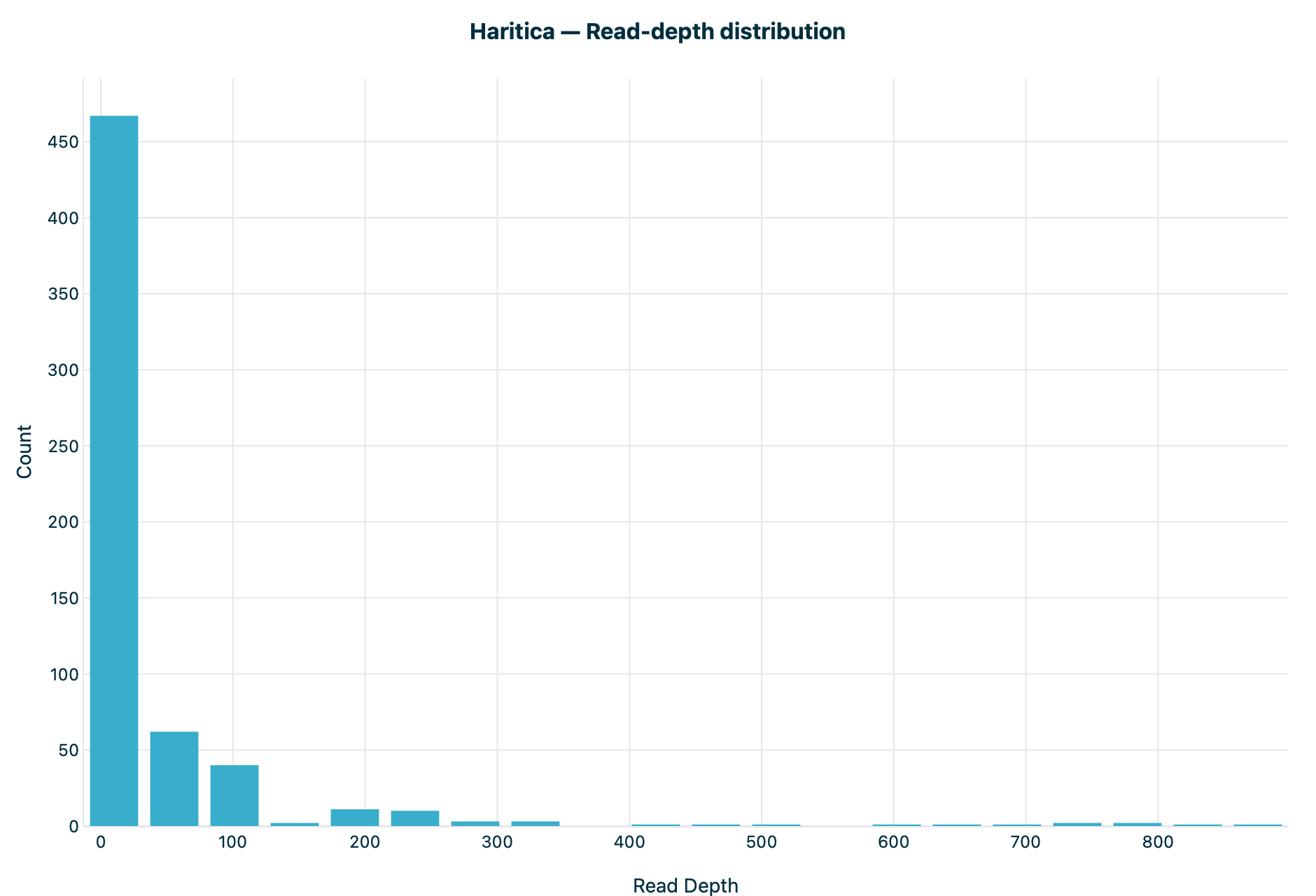
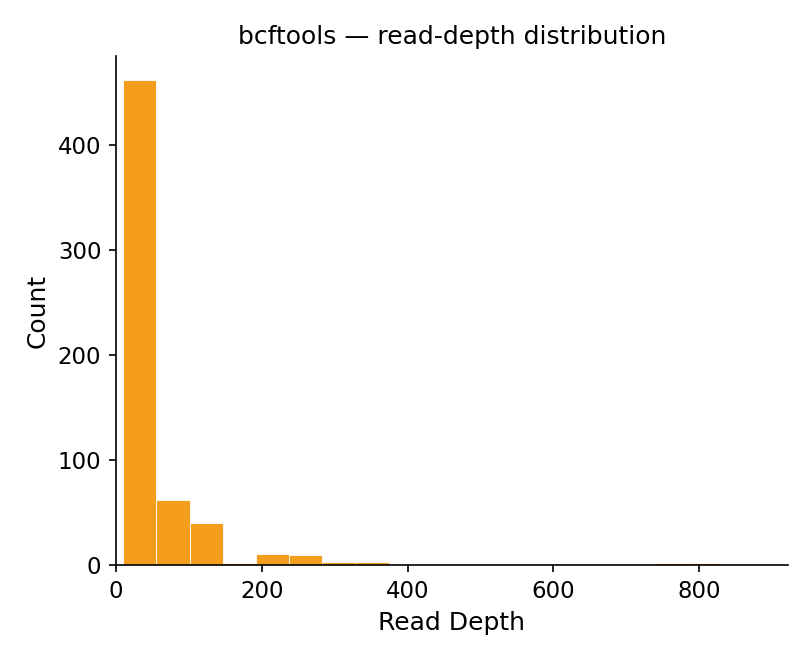
Figure 2. Read-depth distribution. Both are count versus read depth on
the same 0–900× axis, with a sharp mode at low depth (QC floor ≥10×) and a peak of about
465 variants in the lowest bin.
Haritica
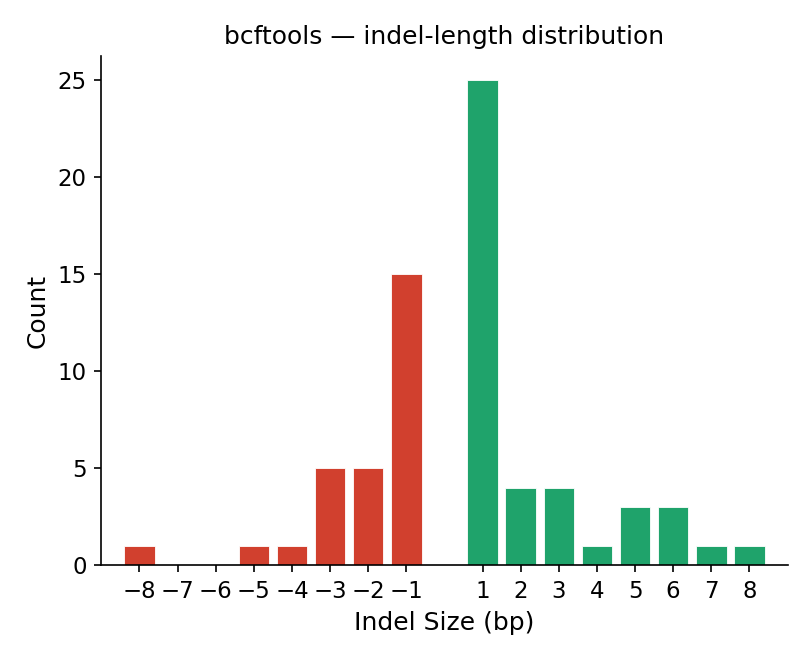
bcftools stats (IDD) — reference
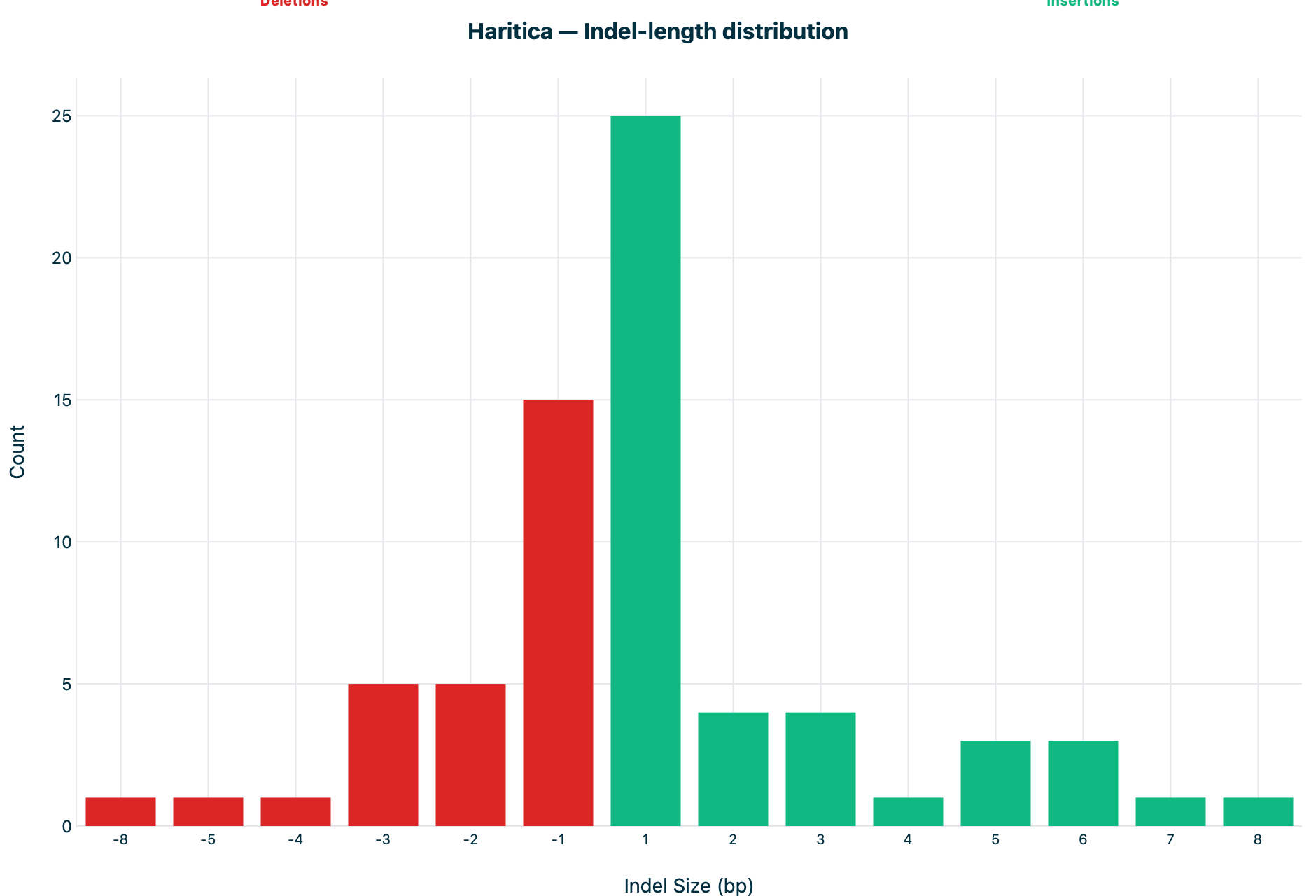
Figure 3. Indel-length distribution. Count versus indel size (bp) on the
same −8…8 axis (deletions red, insertions green); the +1 bp peak is 25 and the
−1 bp peak is 15 on both.
Haritica
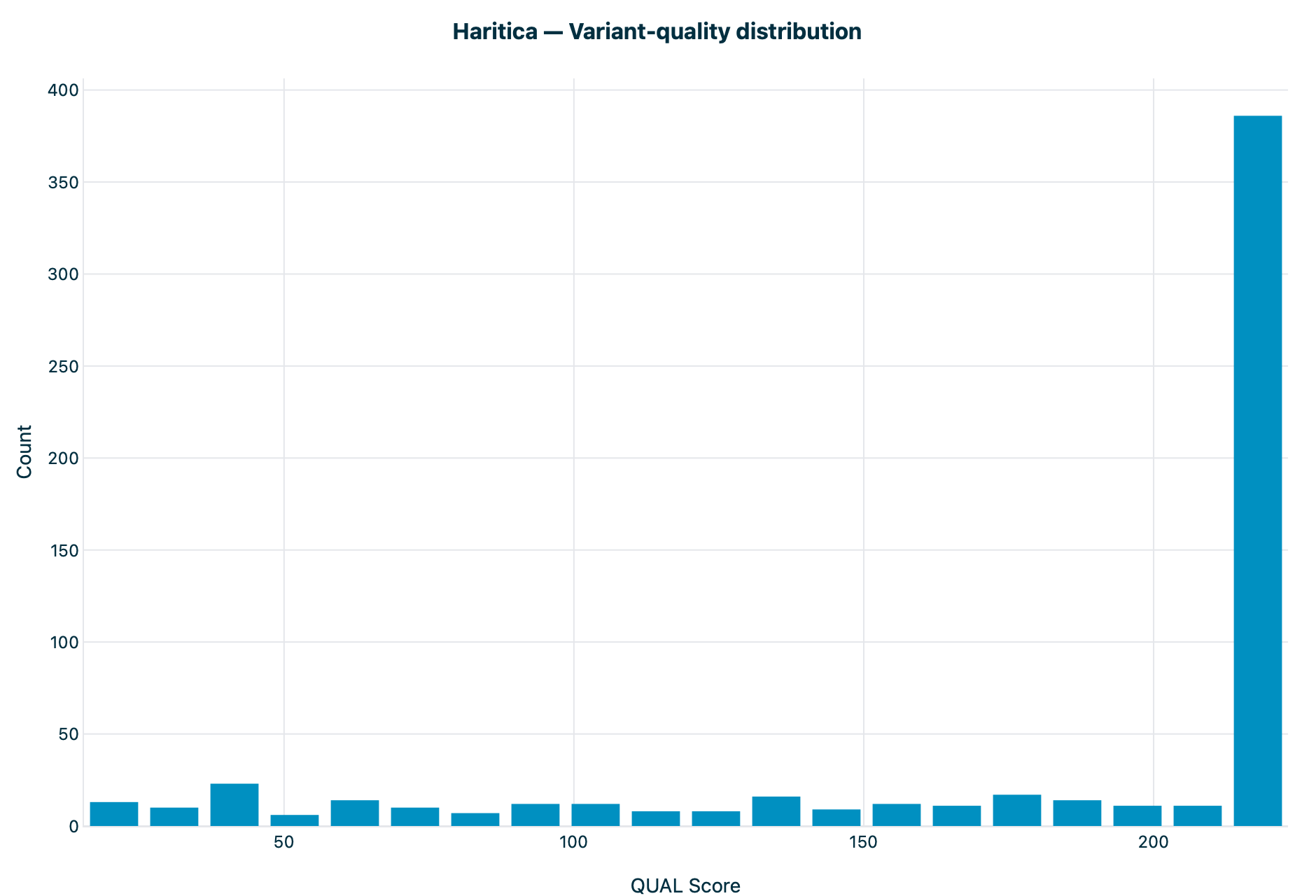
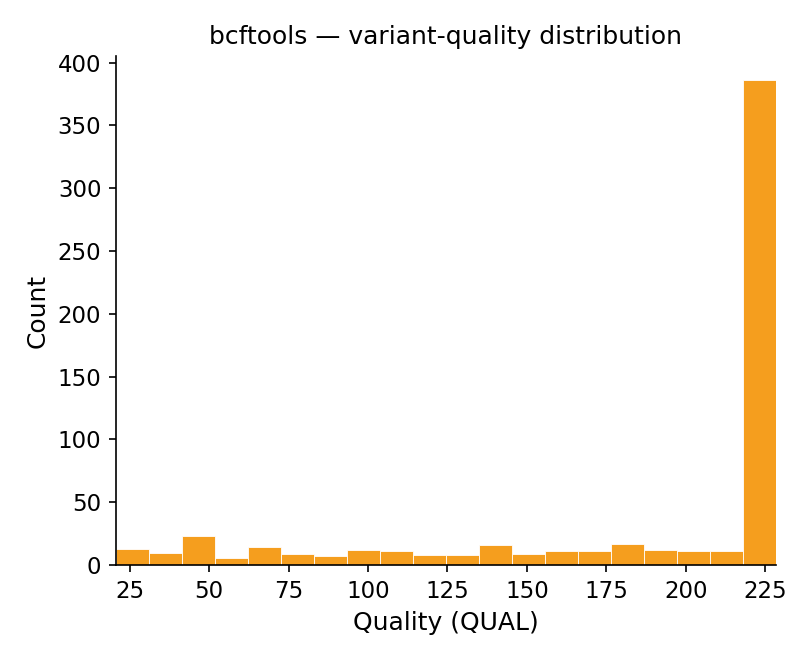
bcftools stats (QUAL) — reference
Figure 4. Variant-quality distribution. Count versus QUAL on the same
~20–225 axis; a flat low body with the same 386-variant pile at the high-quality ceiling on
both.
2.1 Impact and consequence distributions (annotation, app only)
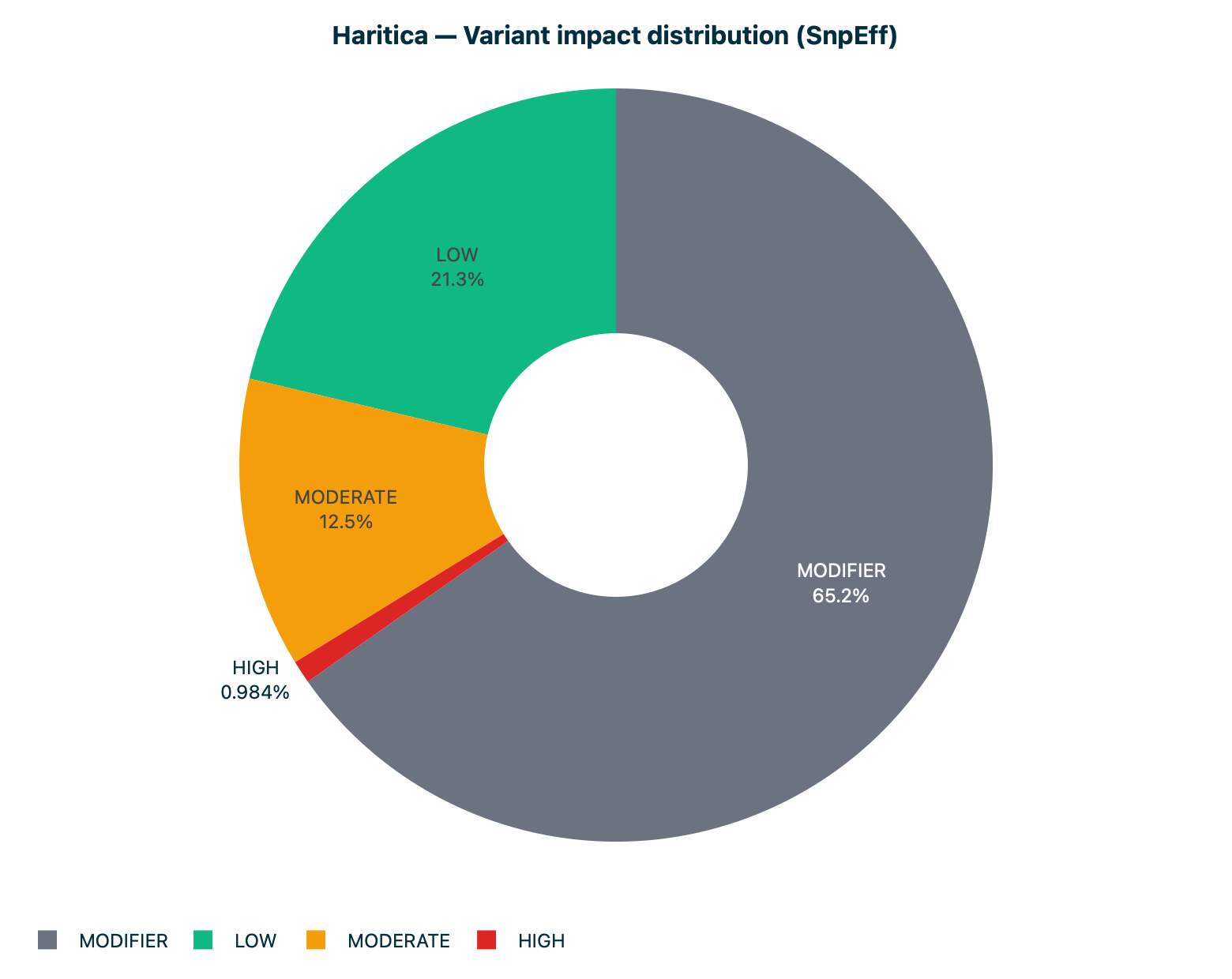
The impact donut (Figure 5) renders SnpEff's impact tiers on the 610 quality-passed variants, and the
consequence bar (Figure 6) renders SnpEff's effect table. SnpEff's native charts depend on the legacy
Google Charts API, so there is no permissive plot to overlay; the annotation engine is validated instead
against bcftools csq at 66.8% coarse-class agreement (§4). Tables 2 and 3 give the
underlying counts.
Figure 5. Impact distribution. SnpEff's four impact tiers over the
610 quality-passed variants (Table 2).
Table 2. SnpEff impact tiers over the 610 quality-passed variants.
Impact
Count
%
MODIFIER
398
65.2
LOW
130
21.3
MODERATE
76
12.5
HIGH
6
1.0
Total
610
100
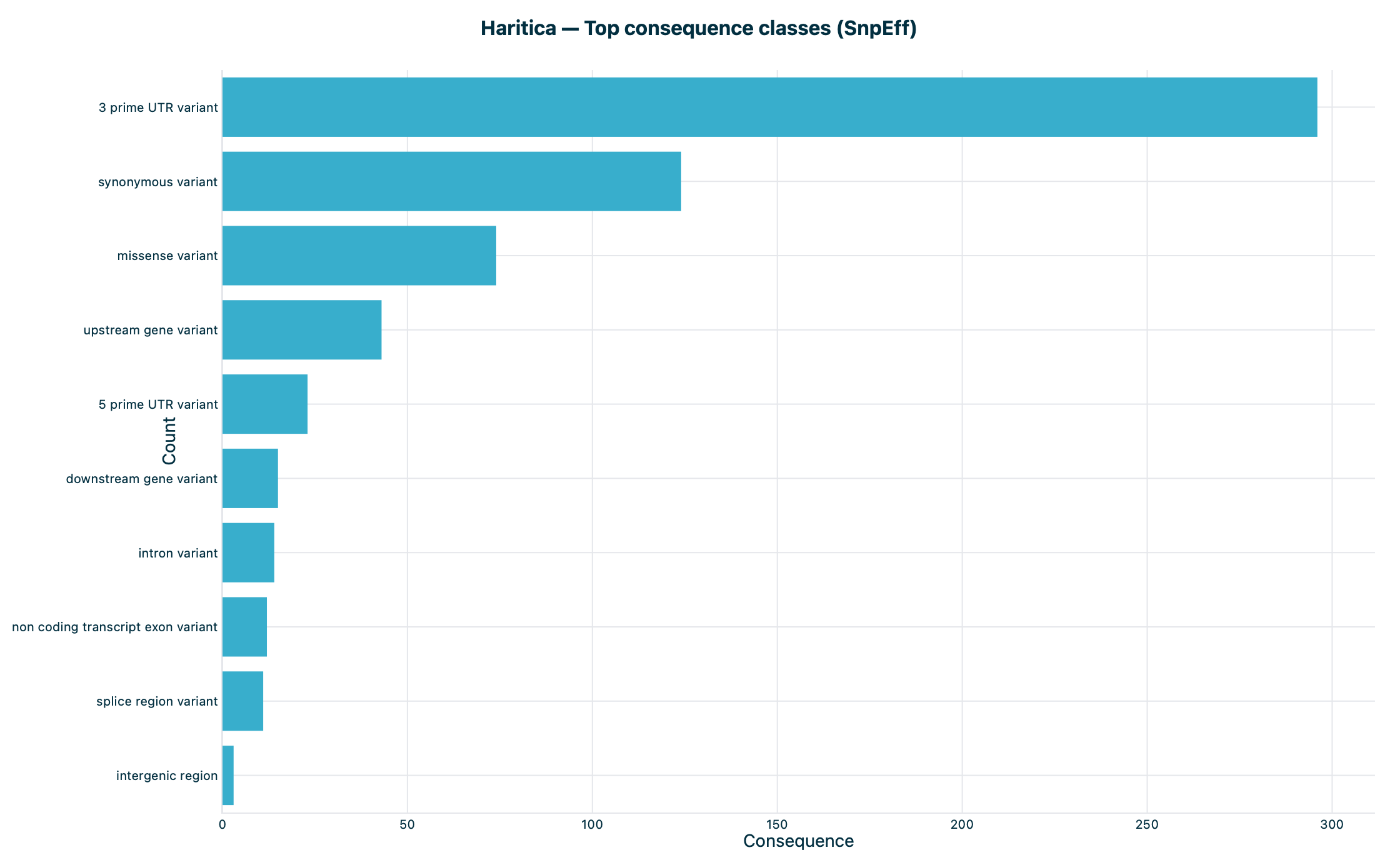
Figure 6. Top consequences. SnpEff's most frequent effect classes over
the quality-passed variants (Table 3).
Table 3. Top SnpEff consequence classes over the quality-passed variants.
Consequence (SnpEff)
Count
3′ UTR variant
296
synonymous variant
124
missense variant
74
upstream gene variant
43
5′ UTR variant
23
downstream gene variant
15
intron variant
14
splice region variant
11
frameshift variant
2
Haritica
Haritica
Figure 7. Oncoplot and rainfall (app only). maftools-style
single-sample visualizations shown for completeness; maftools (GPL R) is the only canonical source and is
excluded for licensing, so no permissive reference is overlaid.
3Calling validation
Haritica's variant statistics reproduce bcftools stats bit-for-bit: the Ti/Tv ratio, the
transition and transversion totals, and the SNV count are identical, as is the full 12-class spectrum
(Figure 1). The calls themselves are scored against the NIST GIAB DNA truth set on chr20/21 within the
benchmark's high-confidence regions. Of 500 evaluable calls, 265 are true positives and 235 are false
positives, giving precision 0.53; excluding the ADAR A>G / T>C RNA-editing signature — real RNA
variants absent from a DNA truth set — raises precision to 0.62. A>G / T>C substitutions account
for 31.1% of the false positives, consistent with that signature. Region-level recall is low by construction
(RNA-seq samples only expressed exons of a whole-genome DNA truth), so precision is the informative metric;
moderate precision is expected for bcftools RNA-seq calling without RNA-aware filtering.
Table 4. Statistics parity: Haritica versus bcftools on the identical VCF.
Quantity
bcftools
Haritica
Ti/Tv ratio
3.06
3.06
Transitions
407
407
Transversions
133
133
Total SNVs
540
540
Indels
70
70
Table 5. Accuracy versus the NIST GIAB HG001 truth set (chr20/21, high-confidence regions).
Metric
Value
True positives (in GIAB truth)
265
False positives (not in DNA truth)
235
Precision
0.530
Precision (excl. RNA-editing FPs)
0.621
A>G / T>C among FPs (ADAR signature)
31.1%
4Annotation validation
SnpEff's consequence calls are compared per variant against bcftools csq on the identical
VCF, both vocabularies mapped to coarse Sequence Ontology classes over the 2,393 variants both tools
annotate (Table 6). Coarse-class agreement is 1,599 / 2,393 = 66.8%, rising to 97.1%
once the two gene-proximal categories bcftools csq structurally cannot emit
(upstream_gene_variant, downstream_gene_variant) are set aside. On the consequences
that matter for interpreting RNA-seq variants — coding and UTR — the two tools agree almost
exactly: UTR 801/811, synonymous 196/197, missense 160/168, splice 40/45, frameshift 5/5, stop/start 4/4.
The residual disagreement is concentrated in one row. bcftools csq annotates only variants
inside a transcript (or at a splice site) and has no near-a-gene vocabulary, so a variant SnpEff labels as
gene-proximal that happens to fall inside some transcript is labelled intron by csq; this
accounts for most of csq's higher intron count (1,031 versus 362). The difference is one-directional and is
a labelling difference, not a calling error: of SnpEff's 362 introns, csq agrees on 355. SnpEff and
bcftools csq also use Ensembl annotations eleven releases apart (GRCh38.99 versus release 110),
so a position just outside a gene in the older model can sit inside a lengthened intron in the newer one.
SnpEff assigns impact tiers that bcftools csq does not (MODIFIER 2,437, LOW 241,
MODERATE 168, HIGH 28), and reported no chromosome-naming warnings.
Table 6. Per-variant consequence concordance: SnpEff (Haritica)
versus bcftools csq, mapped to coarse Sequence Ontology classes over the 2,393 shared
variants.
Coarse consequence class
SnpEff
bcftools csq
Agree
UTR (3′ / 5′)
811
829
801
intron
362
1,031
355
upstream gene
495
0
0
downstream gene
252
0
0
synonymous
197
202
196
missense / inframe
168
169
160
splice
56
45
40
non-coding transcript
43
108
38
frameshift
5
5
5
stop / start
4
4
4
Total (shared)
2,393
2,393
1,599
5Data availability and references
All inputs and external references are public. Raw reads: ENA run
SRR14253270 (GM12878 / HG001). Truth set:
NIST Genome-in-a-Bottle HG001 GRCh38 v4.2.1 small-variant benchmark
(NISTv4.2.1 GRCh38 release;
program page). Reference genome: Ensembl
GRCh38 (release 110), chromosomes 20 and 21. Reference tools, called but not implemented:
bcftools and
SnpEff. The independent concordance reference is generated by
reference_concordance.py and the annotation comparison by
annotation_concordance.py.
Zook JM, McDaniel J, Olson ND, et al. An open resource for accurately benchmarking small variant and reference calls. Nature Biotechnology 2019;37:561–566. (NIST Genome in a Bottle, HG001 / NA12878.)
Danecek P, Bonfield JK, Liddle J, et al. Twelve years of SAMtools and BCFtools. GigaScience 2021;10(2):giab008.
Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012;6(2):80–92.